SMA Kinematics - Treated Before vs After Symptoms
2025-08-11
Read in the data
## load in required data
SMA_DAT <- read.csv("~/Library/CloudStorage/Box-Box/CLA RSS Data Sharing/1022295_McGrattan_SMA_KINEMATICS/SMA_Data/SMA_DAT_summary_last20250811.csv", header = T)
SMA_DAT_full <- read.csv("~/Library/CloudStorage/Box-Box/CLA RSS Data Sharing/1022295_McGrattan_SMA_KINEMATICS/SMA_Data/SMA_DAT_combined_last20250811.csv")
#load in included and last exam data
load("~/Library/CloudStorage/Box-Box/CLA RSS Data Sharing/577565_McGrattan_SMA/BABYVFSSIMP/data/SMA_analysis_files.Rdata")
#Participants NZ_01 and PRI_O4 only have one observation/swallow, so removing them from the analysis; also remove those who are not on inclusion list. Also remove untreated group
SMA_DAT <- SMA_DAT %>%
filter(!(Subject.ID %in% c("NZ_01", "PRI_04"))) %>%
filter(!is.na(treat_group))
SMA_DAT_full <- SMA_DAT_full %>%
filter(!(Subject.ID %in% c("NZ_01", "PRI_04"))) %>%
filter(!is.na(treat_group))
#add in Secretion, Respiration, and FOIS impairments into the data
funct_impair <- analysis_included_lastexam %>%
select(Subject.ID, fois_imp, fois_imp_n, resp_text_any, resp_imp_n, sec_imp, sec_imp_n) Who is missing from the kinematics data?
analysis_included_lastexam %>%
filter(Subject.ID %in% SMA_DAT$Subject.ID == FALSE) %>%
select(Subject.ID, studyid_clean, treat_group) %>%
kable()
SMA_DAT <- SMA_DAT %>%
inner_join(funct_impair, by="Subject.ID")Total Sample N
For the BABYVFSSIMP, there were two analyses - one for untreated participants and the other comparing groups who were treated before vs after symptoms appeared. This analysis just looks at the treated before vs after symptoms. This uses data from the last exam.
## number of infants in treated and untreated groups
visit_summary <- SMA_DAT %>%
filter(!is.na(treat_group)) %>%
group_by(treat_group) %>%
summarize(avg_obs = mean(n_obvs),
min_obs = min(n_obvs),
max_obs = max(n_obvs),
N = n_distinct(Subject.ID))
visit_summary %>%
kable(col.names = c("Treated", "Average Swallows", "Min Swallows", "Max Swallows", "N individuals"),
digits = 2)| Treated | Average Swallows | Min Swallows | Max Swallows | N individuals |
|---|---|---|---|---|
| After Symptoms | 23.39 | 1 | 127 | 51 |
| Before Symptoms | 24.67 | 2 | 61 | 17 |
PCR cleaning
Who in Before has PCR >= 0.5?
Any swallow:
SMA_DAT_full %>%
filter(Measurement_name == "PCR", value >= 0.5, treat_group == "Before Symptoms") %>%
select(Subject.ID, Participant_Num, Swallow_Number, Measurement_name, value) %>%
arrange(Subject.ID, Participant_Num, Swallow_Number) %>%
kable()Worst/best/mean:
SMA_DAT %>%
filter(Measurement_name == "PCR", treat_group == "Before Symptoms") %>%
filter(Mean >= 0.5 | Worst_OI >= 0.5 | Best_OI >= 0.5) %>%
select(Subject.ID, Measurement_name, Mean, Worst_OI, Best_OI) %>%
arrange(Subject.ID) %>%
kable()Align OI scores for OI_17 (tongue base), OI_19 , and OI_20 (residue) with worst PCR scores.
pcrworst <- SMA_DAT %>%
filter(Measurement_name == "PCR") %>%
select(Subject.ID, Worst_OI, treat_group) %>%
rename(PCR_Worst_OI = Worst_OI)
analysis_included_lastexam %>%
select(Subject.ID, oi_17, oi_19, oi_20) %>%
right_join(pcrworst, by="Subject.ID") %>%
relocate(treat_group, .after = Subject.ID) %>%
arrange(treat_group, PCR_Worst_OI) %>%
kable()Descriptive Statistics
Overall descriptives
First, take averages across all swallows in each participants.
SMA_DAT_summary_indiv <- SMA_DAT_full %>%
group_by(Measurement_name, treat_group, Subject.ID) %>%
summarize(Mean = mean(value, na.rm=T),
SD = sd(value, na.rm=T),
N = n())
datatable(SMA_DAT_summary_indiv, options = list(pageLength = 10))Then take the mean and SD of these means
SMA_DAT_summary_indiv %>%
group_by(Measurement_name, treat_group) %>%
summarize(Overall_Mean = mean(Mean, na.rm=T),
Overall_SD = sd(Mean, na.rm=T),
N = n()) %>%
kbl(digits=2) %>%
kable_styling()| Measurement_name | treat_group | Overall_Mean | Overall_SD | N |
|---|---|---|---|---|
| HPT | After Symptoms | 1.01 | 0.99 | 51 |
| HPT | Before Symptoms | 0.76 | 0.74 | 17 |
| OPT | After Symptoms | 0.78 | 0.62 | 51 |
| OPT | Before Symptoms | 0.95 | 1.18 | 17 |
| PCR | After Symptoms | 0.15 | 0.16 | 51 |
| PCR | Before Symptoms | 0.03 | 0.02 | 17 |
| PESop | After Symptoms | 0.24 | 0.09 | 51 |
| PESop | Before Symptoms | 0.22 | 0.06 | 17 |
| TPT | After Symptoms | 1.74 | 1.40 | 51 |
| TPT | Before Symptoms | 1.72 | 1.87 | 17 |
Visualizations
SMA_DAT_l <- SMA_DAT %>%
select(Subject.ID,treat_group, Measurement_name, Worst_OI, Best_OI, Mean) %>%
pivot_longer(cols = Worst_OI:Mean,
names_to = "Statistic",
values_to = "Value")
SMA_DAT_l %>%
filter(Statistic == "Mean") %>%
ggplot(aes(x = treat_group, y=Value)) +
geom_boxplot() +
facet_wrap(~Measurement_name, scales = "free") +
labs(title = "Mean") +
theme_classic() +
theme(axis.text.x = element_text(angle = 90))## Warning: Removed 14 rows containing non-finite outside the scale range
## (`stat_boxplot()`).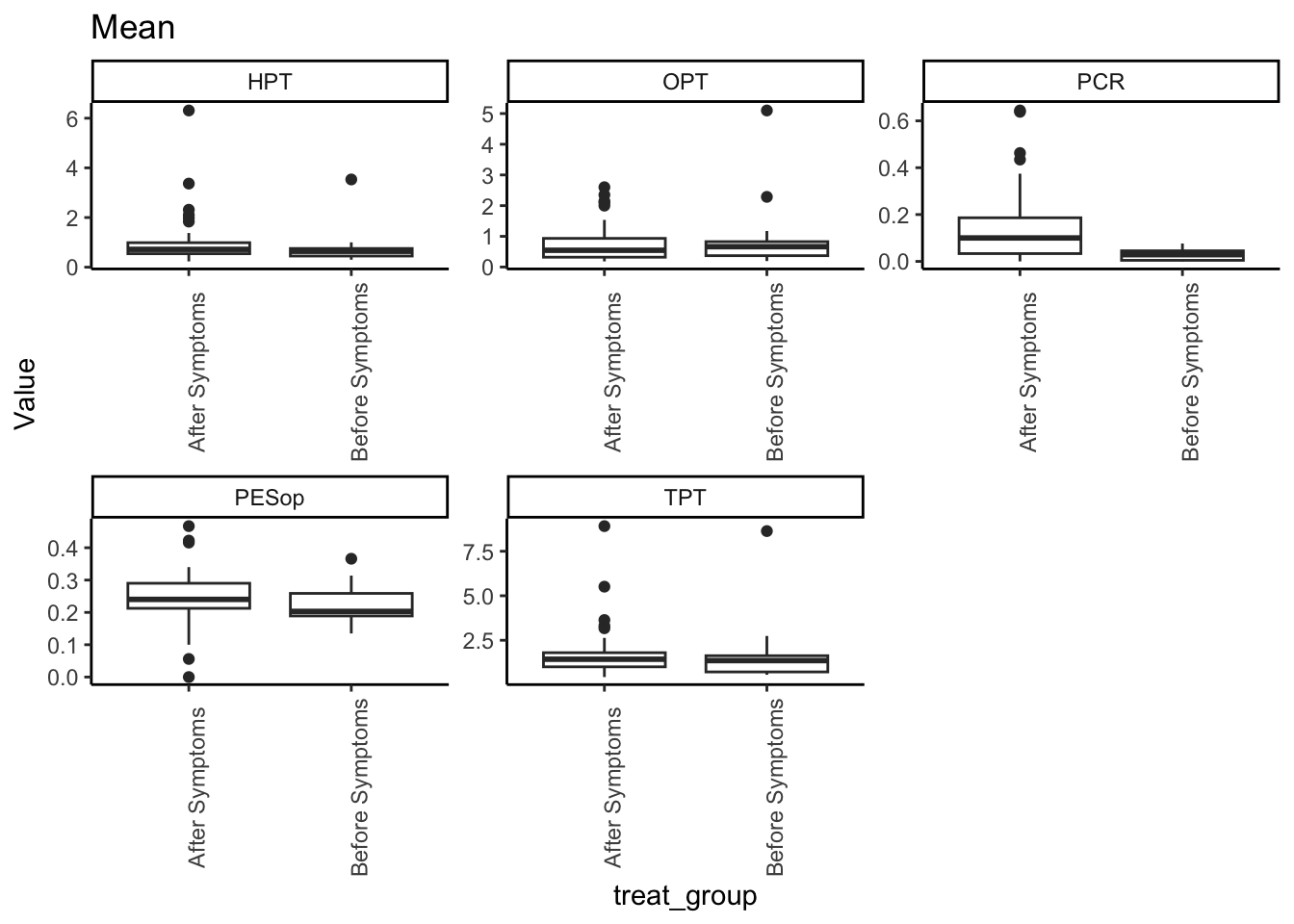
Do OI scores differ for by treatment?
These are continuous measures, although groups are slightly unbalanced in terms of numbers. Will use parametric comparisons for now. t-tests will be used to asses differences between means for each measure.
PCR - Asymp vs Symp
PCR - Mean Score
Means and SD
SMA_DAT %>%
filter(Measurement_name == "PCR") %>%
group_by(treat_group) %>%
summarize(Average = mean(Mean, na.rm=T),
SD = sd(Mean, na.rm=T),
N = sum(!is.na(Mean))) %>%
kable(digits=2)| treat_group | Average | SD | N |
|---|---|---|---|
| After Symptoms | 0.15 | 0.16 | 49 |
| Before Symptoms | 0.03 | 0.02 | 17 |
#look at the raw distributions
SMA_DAT %>%
filter(Measurement_name == "PCR") %>%
ggplot(aes(x = treat_group, y=Mean)) +
geom_boxplot() +
theme_classic()## Warning: Removed 2 rows containing non-finite outside the scale range
## (`stat_boxplot()`).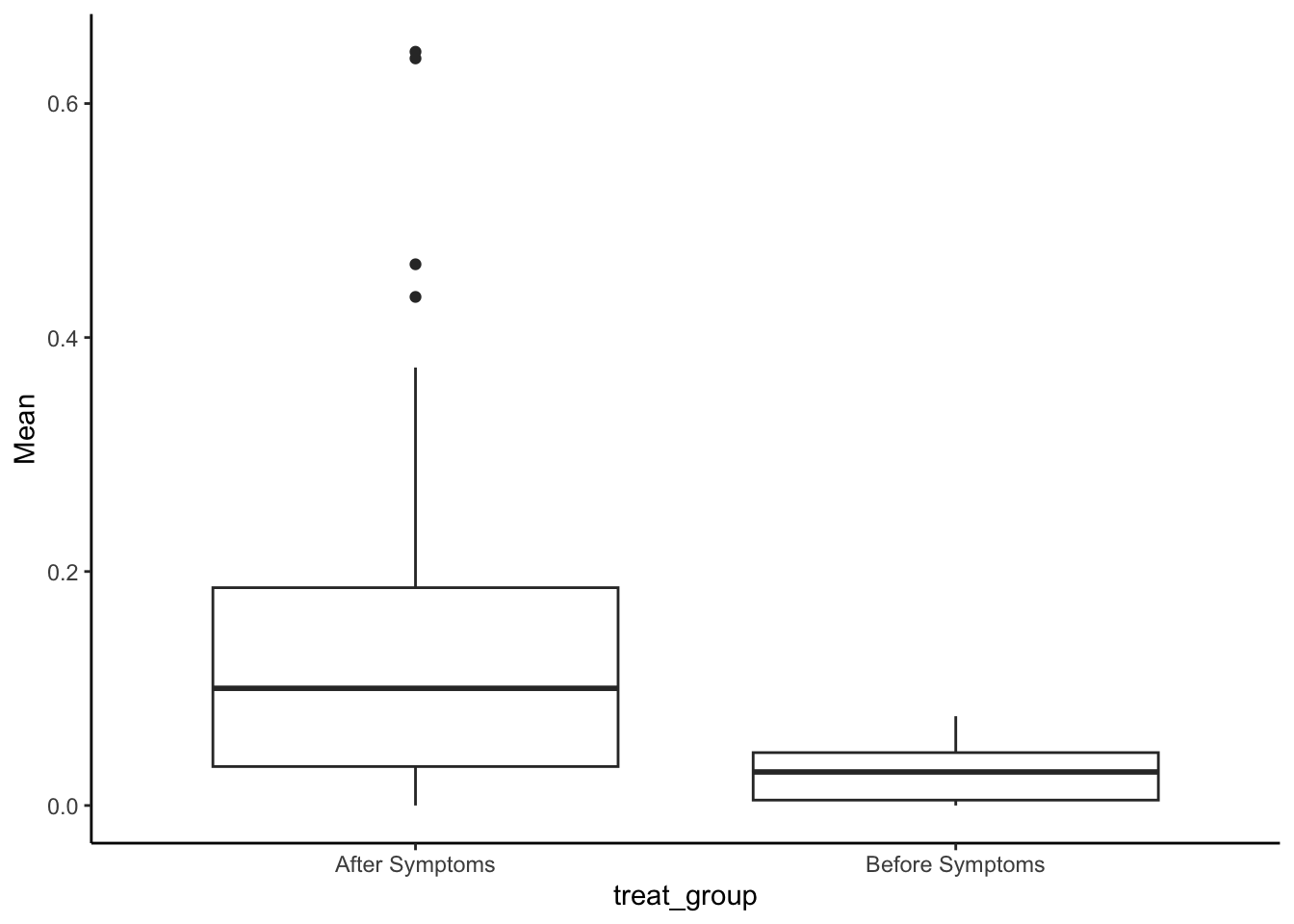
#t.test
(pcr_m <- t.test(Mean ~ treat_group, data = filter(SMA_DAT, Measurement_name == "PCR")))##
## Welch Two Sample t-test
##
## data: Mean by treat_group
## t = 5.1029, df = 53.801, p-value = 4.48e-06
## alternative hypothesis: true difference in means between group After Symptoms and group Before Symptoms is not equal to 0
## 95 percent confidence interval:
## 0.07121631 0.16340334
## sample estimates:
## mean in group After Symptoms mean in group Before Symptoms
## 0.14582697 0.02851715#cohen's d for effect size
cohen.d(Mean ~ treat_group, data = filter(SMA_DAT, Measurement_name == "PCR"))## Call: cohen.d(x = Mean ~ treat_group, data = filter(SMA_DAT, Measurement_name ==
## "PCR"))
## Cohen d statistic of difference between two means
## lower effect upper
## Mean -1.45 -0.88 -0.3
##
## Multivariate (Mahalanobis) distance between groups
## [1] 0.88
## r equivalent of difference between two means
## Mean
## -0.36Dichotimized PCR score
Could you dichotomize average PCR for each kid into <0.2 and >0.2 (profound impairment), give N% for the kids with each in pre and post symptomatic groups, and compare those proportions between groups?
pcr_group <- SMA_DAT %>%
filter(Measurement_name == "PCR") %>%
mutate(pcrimp = ifelse(Mean <= 0.2, "No", "Yes"))
pcr_group %>%
filter(!is.na(pcrimp)) %>%
group_by(treat_group, pcrimp) %>%
tally() %>%
group_by(treat_group) %>%
mutate(percent = n/sum(n)*100) %>%
kable(digits = 1)| treat_group | pcrimp | n | percent |
|---|---|---|---|
| After Symptoms | No | 37 | 75.5 |
| After Symptoms | Yes | 12 | 24.5 |
| Before Symptoms | No | 17 | 100.0 |
pcrimptab <- table( pcr_group$pcrimp, pcr_group$treat_group)
fisher.test(pcrimptab)##
## Fisher's Exact Test for Count Data
##
## data: pcrimptab
## p-value = 0.02742
## alternative hypothesis: true odds ratio is not equal to 1
## 95 percent confidence interval:
## 0.0000000 0.9084527
## sample estimates:
## odds ratio
## 0PESop - Asymp vs Symp
PESop - Mean Score
Means and SD
SMA_DAT %>%
filter(Measurement_name == "PESop") %>%
group_by(treat_group) %>%
summarize(Average = mean(Mean, na.rm=T),
SD = sd(Mean, na.rm=T),
N = sum(!is.na(Mean))) %>%
kable(digits=2)| treat_group | Average | SD | N |
|---|---|---|---|
| After Symptoms | 0.24 | 0.09 | 48 |
| Before Symptoms | 0.22 | 0.06 | 17 |
#look at the raw distributions
SMA_DAT %>%
filter(Measurement_name == "PESop") %>%
ggplot(aes(x = treat_group, y=Mean)) +
geom_boxplot() +
theme_classic()## Warning: Removed 3 rows containing non-finite outside the scale range
## (`stat_boxplot()`).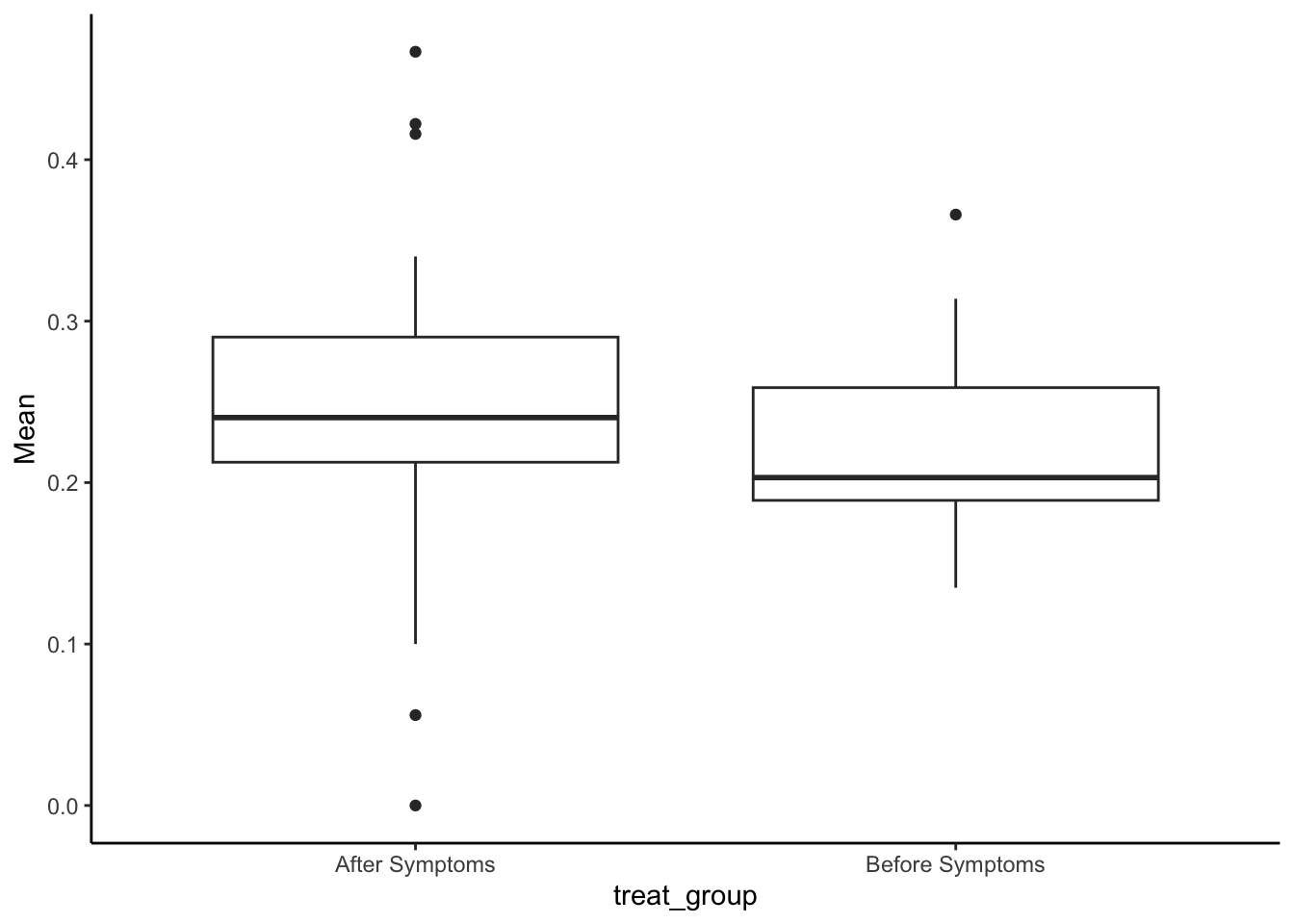
#t.test
(pesop_m <- t.test(Mean ~ treat_group, data = filter(SMA_DAT, Measurement_name == "PESop")))##
## Welch Two Sample t-test
##
## data: Mean by treat_group
## t = 0.84042, df = 40.039, p-value = 0.4057
## alternative hypothesis: true difference in means between group After Symptoms and group Before Symptoms is not equal to 0
## 95 percent confidence interval:
## -0.02343201 0.05679291
## sample estimates:
## mean in group After Symptoms mean in group Before Symptoms
## 0.2399721 0.2232917#cohen's d
cohen.d(Mean ~ treat_group, data = filter(SMA_DAT, Measurement_name == "PESop"))## Call: cohen.d(x = Mean ~ treat_group, data = filter(SMA_DAT, Measurement_name ==
## "PESop"))
## Cohen d statistic of difference between two means
## lower effect upper
## Mean -0.76 -0.2 0.35
##
## Multivariate (Mahalanobis) distance between groups
## [1] 0.2
## r equivalent of difference between two means
## Mean
## -0.09Does PCR scores predict functional impairments?
Using analyses similar to the Babyvfssimp scores. Look at point-biserial correlations and logistic regression.
Secreation management
Point-biserial associations :
SMA_DAT %>%
filter(Measurement_name == "PCR") %>%
select(sec_imp_n, Mean) %>%
summarize(mean_corr = cor(sec_imp_n, Mean, use="pairwise.complete.obs"),
n = sum(!is.na(Mean)),
p = cor.test(sec_imp_n, Mean)$p.value) %>%
kable(digits = 2)| mean_corr | n | p |
|---|---|---|
| 0.26 | 66 | 0.03 |
Plots:
SMA_DAT %>%
filter(Measurement_name == "PCR") %>%
ggplot(aes(x = sec_imp_n, y = Mean)) +
geom_point(position = position_jitter(width = .05)) +
geom_smooth(method = "lm", se = F) +
theme_classic()## Warning: Removed 2 rows containing non-finite outside the scale range
## (`stat_smooth()`).## Warning: Removed 2 rows containing missing values or values outside
## the scale range (`geom_point()`).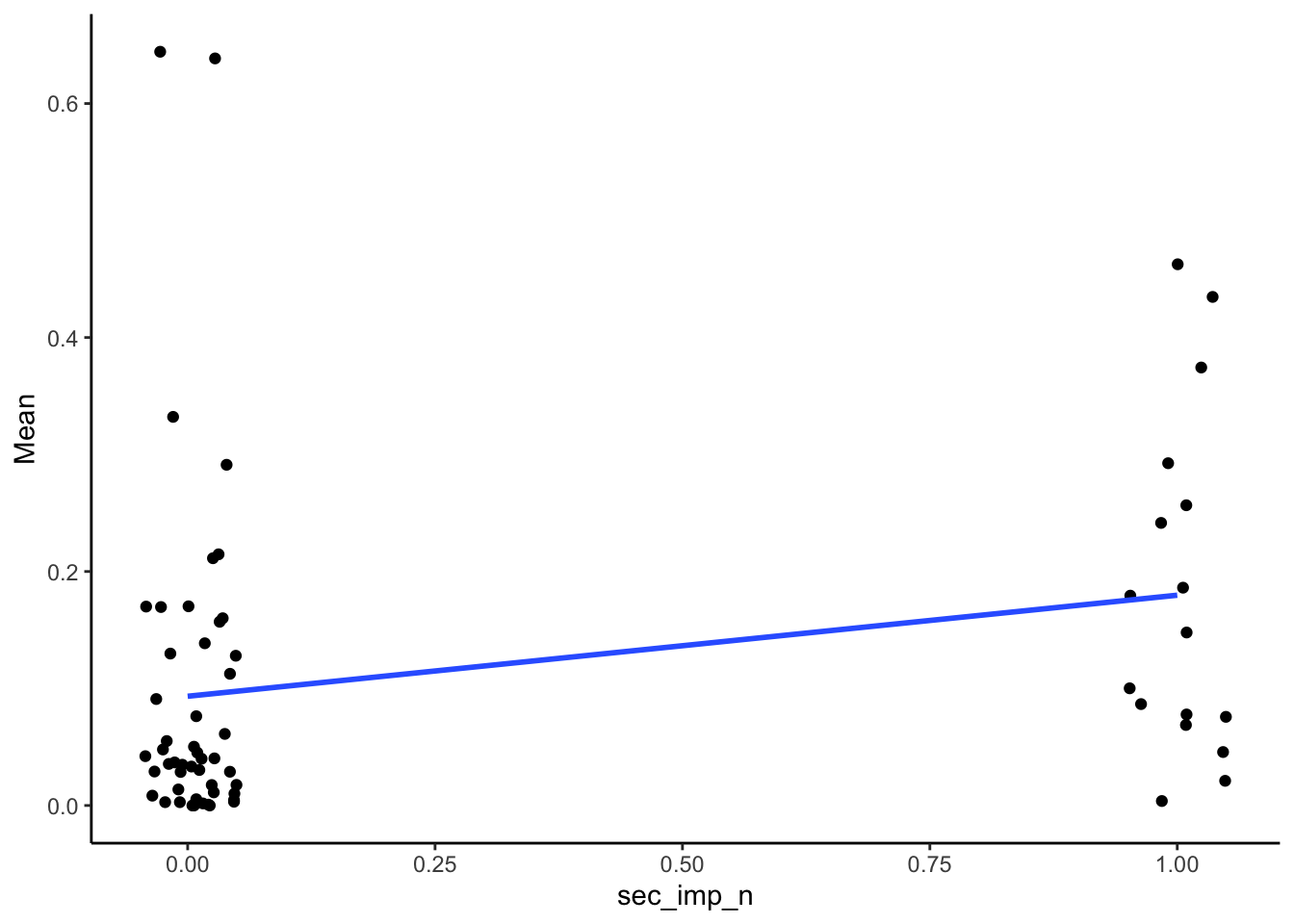
Logistic regressions
secpcr_logr <- glm(sec_imp_n ~ Mean, family = "binomial", data = filter(SMA_DAT, Measurement_name == "PCR"))
secpcr_logr %>%
tidy() %>%
kable(digits = 3)| term | estimate | std.error | statistic | p.value |
|---|---|---|---|---|
| (Intercept) | -1.551 | 0.393 | -3.949 | 0.000 |
| Mean | 3.799 | 1.916 | 1.983 | 0.047 |
summary(secpcr_logr)##
## Call:
## glm(formula = sec_imp_n ~ Mean, family = "binomial", data = filter(SMA_DAT,
## Measurement_name == "PCR"))
##
## Coefficients:
## Estimate Std. Error z value Pr(>|z|)
## (Intercept) -1.5508 0.3927 -3.949 7.84e-05 ***
## Mean 3.7988 1.9161 1.983 0.0474 *
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1
##
## (Dispersion parameter for binomial family taken to be 1)
##
## Null deviance: 75.307 on 65 degrees of freedom
## Residual deviance: 71.131 on 64 degrees of freedom
## (2 observations deleted due to missingness)
## AIC: 75.131
##
## Number of Fisher Scoring iterations: 4Mean PCR significant predicts impairment in secretion management. 3.7987782, the increase in odds for every 1 point increase in PCR (which note the small scale of this value) is 44.6466026
FOIS
Point-biserial associations
SMA_DAT %>%
filter(Measurement_name == "PCR") %>%
summarize(pb_correlation = cor(fois_imp_n, Mean, use="pairwise.complete.obs"),
n = sum(!is.na(Mean)),
p = cor.test(fois_imp_n, Mean)$p.value) %>%
kable(digits = 2)| pb_correlation | n | p |
|---|---|---|
| 0.15 | 66 | 0.23 |
Plots:
SMA_DAT %>%
filter(Measurement_name == "PCR") %>%
ggplot(aes(x = fois_imp_n, y = Mean)) +
geom_point(position = position_jitter(width = .05)) +
geom_smooth(method = "lm", se = F) +
theme_classic()## Warning: Removed 2 rows containing non-finite outside the scale range
## (`stat_smooth()`).## Warning: Removed 2 rows containing missing values or values outside
## the scale range (`geom_point()`).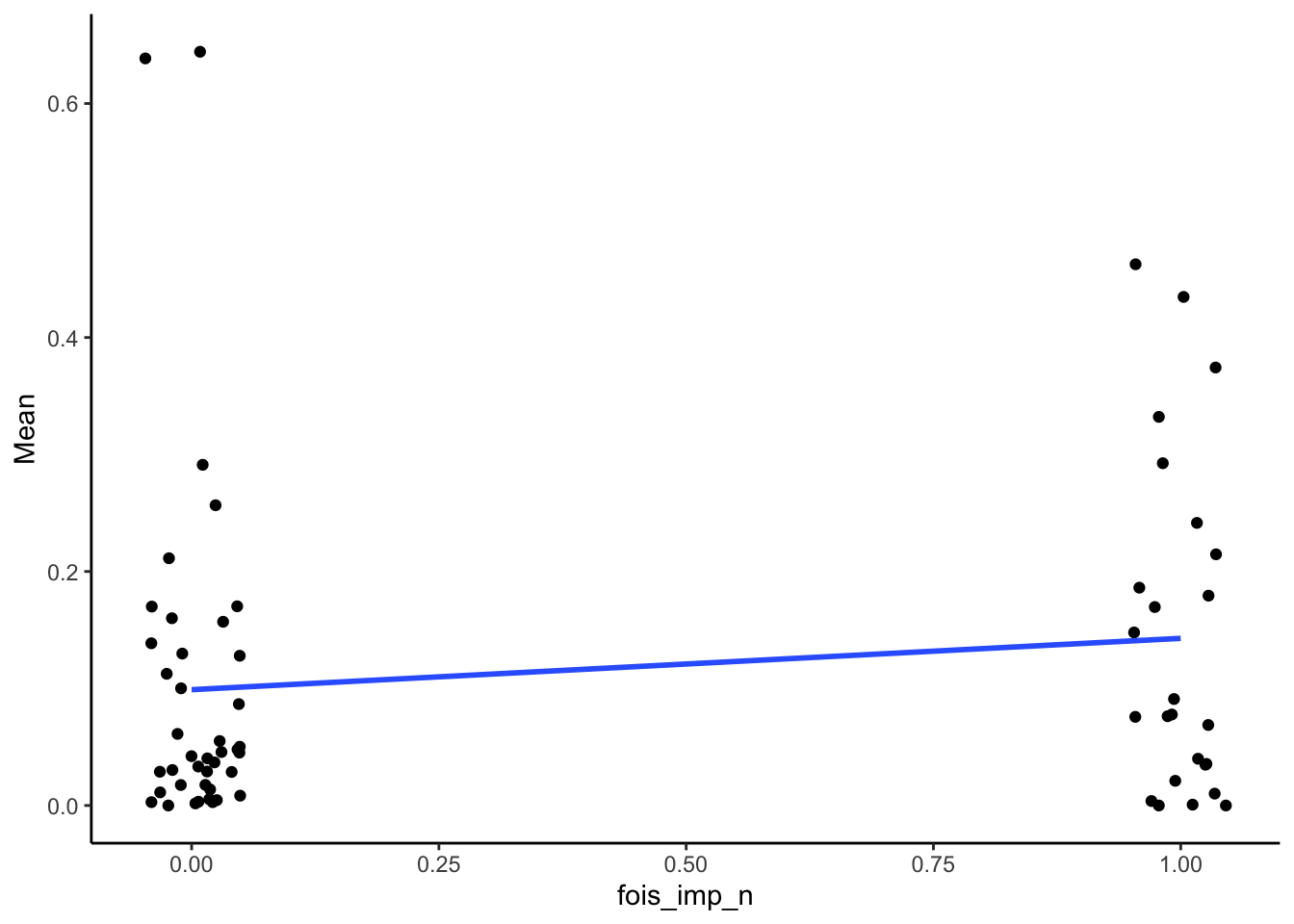
Logistic regressions
foispcr_logr <- glm(fois_imp_n ~ Mean, family = "binomial", data = filter(SMA_DAT, Measurement_name == "PCR"))
foispcr_logr %>%
tidy() %>%
kable(digits = 3)| term | estimate | std.error | statistic | p.value |
|---|---|---|---|---|
| (Intercept) | -0.744 | 0.334 | -2.228 | 0.026 |
| Mean | 2.101 | 1.782 | 1.179 | 0.238 |
NS
Respiratory Support
Point-biserial associations
SMA_DAT %>%
filter(Measurement_name == "PCR") %>%
summarize(pb_correlation = cor(resp_imp_n, Mean, use="pairwise.complete.obs"),
n = sum(!is.na(Mean)),
p = cor.test(resp_imp_n, Mean)$p.value) %>%
kable(digits = 2)| pb_correlation | n | p |
|---|---|---|
| 0.32 | 66 | 0.01 |
Plots:
SMA_DAT %>%
filter(Measurement_name == "PCR") %>%
ggplot(aes(x = resp_imp_n, y = Mean)) +
geom_point(position = position_jitter(width = .05)) +
geom_smooth(method = "lm", se = F) +
theme_classic()## Warning: Removed 2 rows containing non-finite outside the scale range
## (`stat_smooth()`).## Warning: Removed 2 rows containing missing values or values outside
## the scale range (`geom_point()`).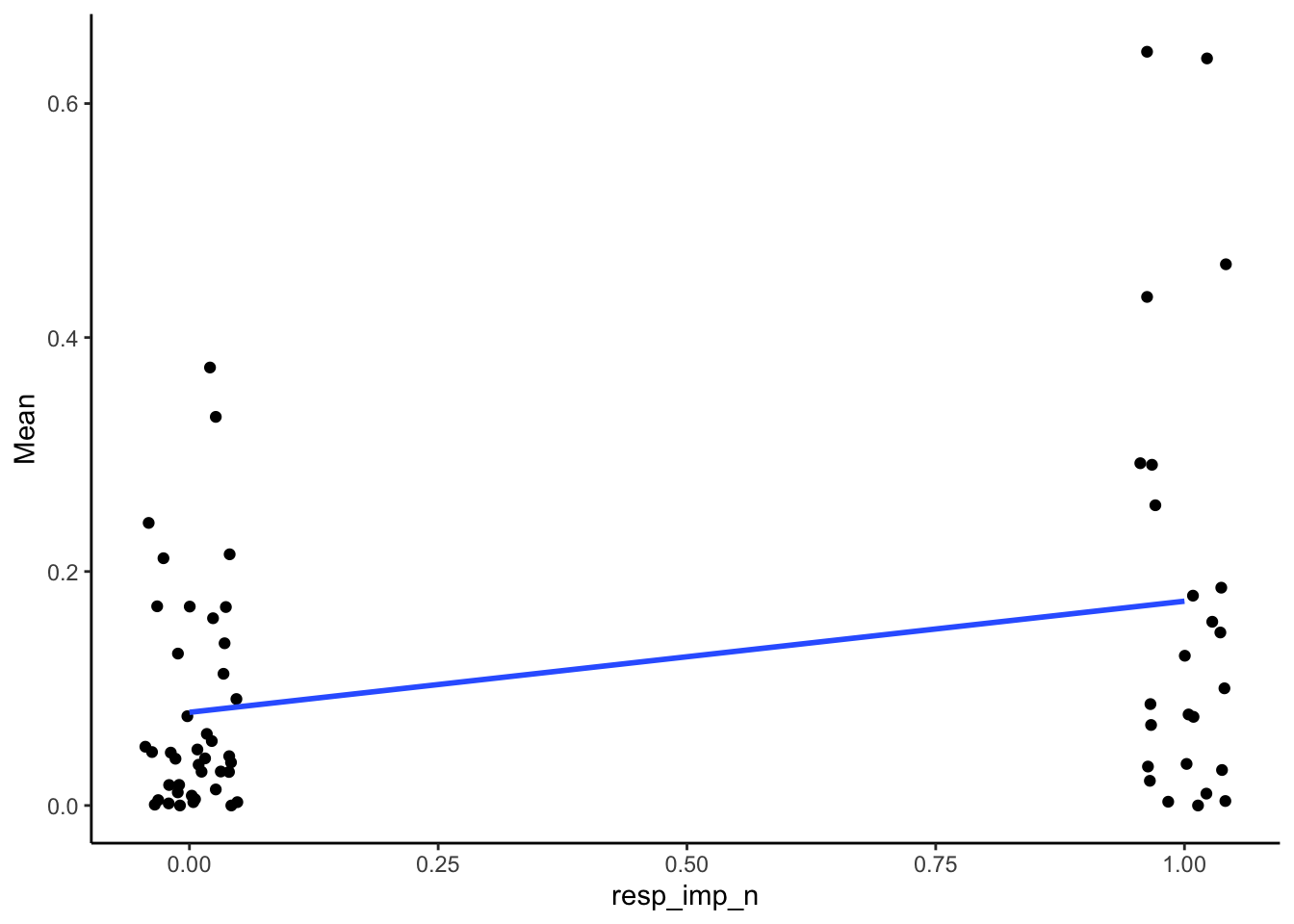
Logistic regressions
resppcr_logr <- glm(resp_imp_n ~ Mean, family = "binomial", data = filter(SMA_DAT, Measurement_name == "PCR"))
resppcr_logr %>%
tidy() %>%
kable(digits = 3)| term | estimate | std.error | statistic | p.value |
|---|---|---|---|---|
| (Intercept) | -1.092 | 0.361 | -3.022 | 0.003 |
| Mean | 5.086 | 2.170 | 2.344 | 0.019 |
Exponentiated effect = 161.6691562.
Comparing PCR and Domain V predictions of Secretion impairment
Putting the datasets together
analysis_included_lastexam_domains <- analysis_included_lastexam %>%
select(Subject.ID, contains("domain"))
SMA_VFSS_combo <- SMA_DAT %>%
filter(Measurement_name == "PCR") %>%
inner_join(analysis_included_lastexam_domains, by="Subject.ID") %>%
filter(!is.na(Mean)) #remove two who have missing PCR dataLook at correlation between PCR and domain V
SMA_VFSS_combo %>%
summarize(pb_correlation = cor(oi_domainV, Mean, use="pairwise.complete.obs"),
n = sum(!is.na(Mean)),
p = cor.test(oi_domainV, Mean)$p.value) %>%
kable(digits = 2)| pb_correlation | n | p |
|---|---|---|
| 0.67 | 66 | 0 |
Very strongly correlated
SMA_VFSS_combo %>%
ggplot(aes(x = oi_domainV, y = Mean)) +
geom_point() +
geom_smooth(method = "lm", se = F) +
theme_classic()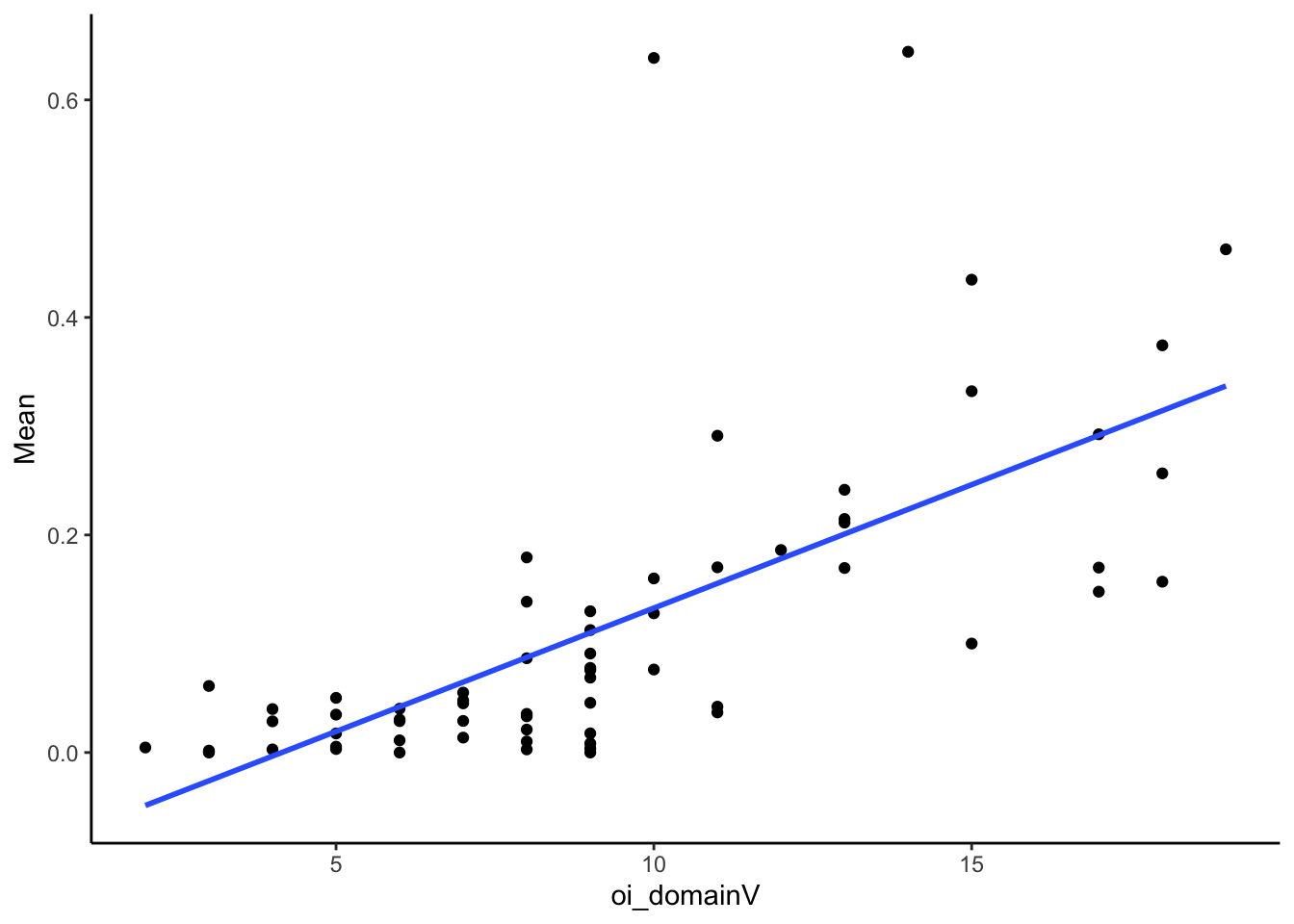
Included in model together? Likely to have multi-colinearity due to high correlation
#start with domain V only (reduced model)
m1_dv_first <- glm(sec_imp_n ~ oi_domainV, family = "binomial", data = SMA_VFSS_combo)
#then add PCR
m2_dv_first <- glm(sec_imp_n ~ oi_domainV + Mean, family = "binomial", data = SMA_VFSS_combo)
#compare
anova(m1_dv_first, m2_dv_first)## Analysis of Deviance Table
##
## Model 1: sec_imp_n ~ oi_domainV
## Model 2: sec_imp_n ~ oi_domainV + Mean
## Resid. Df Resid. Dev Df Deviance Pr(>Chi)
## 1 64 61.275
## 2 63 60.964 1 0.31117 0.577AIC(m1_dv_first, m2_dv_first)## df AIC
## m1_dv_first 2 65.27537
## m2_dv_first 3 66.96420Very little difference between models…
Try the other order (entering Mean first to see if domain score adds more information)
#start with PCR only (reduced model)
m1_pcr_first <- glm(sec_imp_n ~ Mean, family = "binomial", data = SMA_VFSS_combo)
#then add domainV
m2_pcr_first <- glm(sec_imp_n ~ Mean + oi_domainV, family = "binomial", data = SMA_VFSS_combo)
#compare
anova(m1_pcr_first, m2_pcr_first)## Analysis of Deviance Table
##
## Model 1: sec_imp_n ~ Mean
## Model 2: sec_imp_n ~ Mean + oi_domainV
## Resid. Df Resid. Dev Df Deviance Pr(>Chi)
## 1 64 71.131
## 2 63 60.964 1 10.167 0.00143 **
## ---
## Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1AIC(m1_pcr_first, m2_pcr_first)## df AIC
## m1_pcr_first 2 75.13109
## m2_pcr_first 3 66.96420Does PESop scores predict functional impairments?
Using analyses similar to the Babyvfssimp scores. Look at point-biserial correlations and logistic regression.
Secreation management
Point-biserial associations :
SMA_DAT %>%
filter(Measurement_name == "PESop") %>%
select(sec_imp_n, Mean) %>%
summarize(mean_corr = cor(sec_imp_n, Mean, use="pairwise.complete.obs"),
n = sum(!is.na(Mean)),
p = cor.test(sec_imp_n, Mean)$p.value) %>%
kable(digits = 2)| mean_corr | n | p |
|---|---|---|
| -0.14 | 65 | 0.26 |
Plots:
SMA_DAT %>%
filter(Measurement_name == "PESop") %>%
ggplot(aes(x = sec_imp_n, y = Mean)) +
geom_point(position = position_jitter(width = .05)) +
geom_smooth(method = "lm", se = F) +
theme_classic()## Warning: Removed 3 rows containing non-finite outside the scale range
## (`stat_smooth()`).## Warning: Removed 3 rows containing missing values or values outside
## the scale range (`geom_point()`).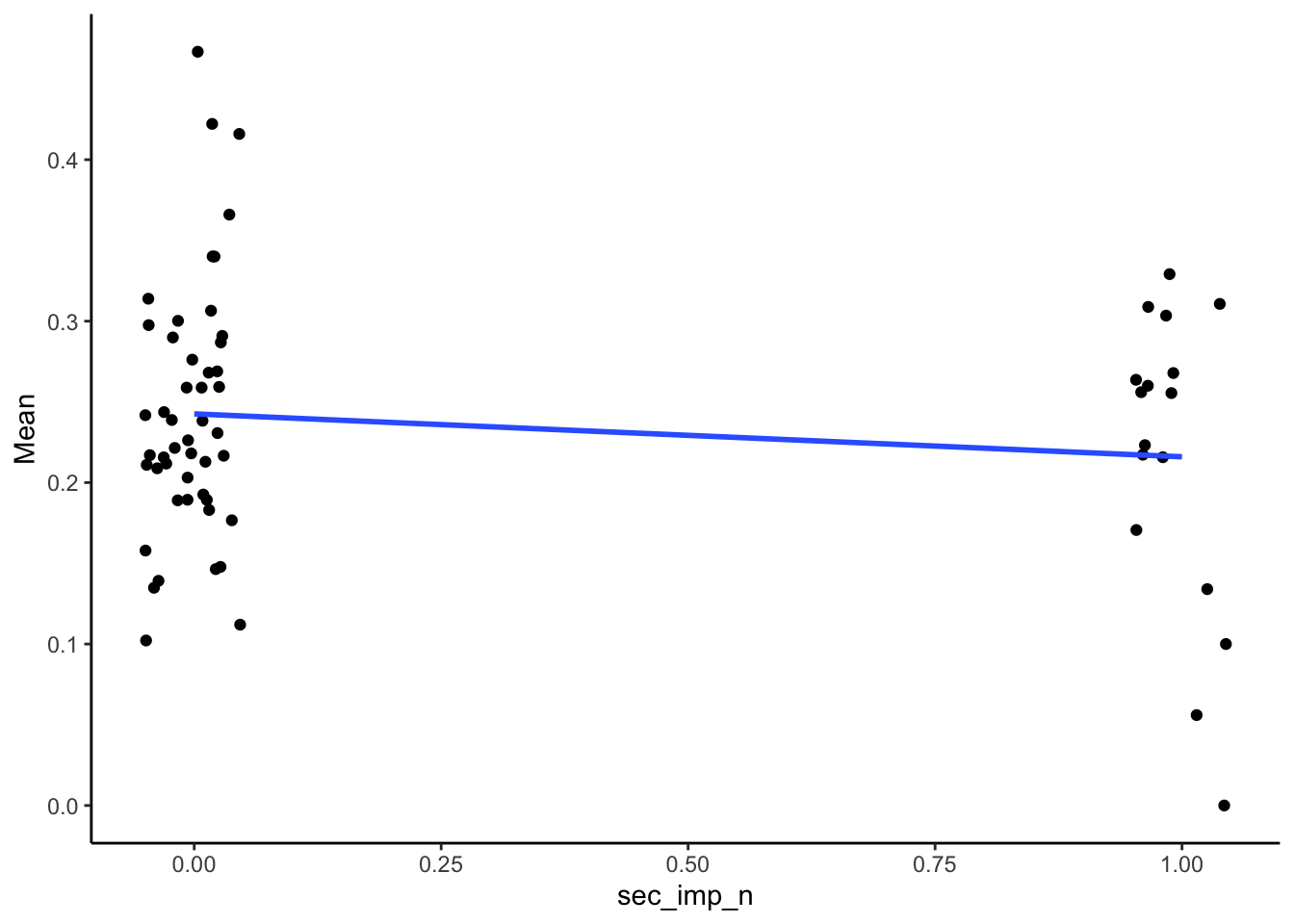
Logistic regressions
secpesop_logr <- glm(sec_imp_n ~ Mean, family = "binomial", data = filter(SMA_DAT, Measurement_name == "PESop"))
secpesop_logr %>%
tidy() %>%
kable(digits = 3)| term | estimate | std.error | statistic | p.value |
|---|---|---|---|---|
| (Intercept) | -0.105 | 0.849 | -0.124 | 0.902 |
| Mean | -4.069 | 3.582 | -1.136 | 0.256 |
NS
FOIS
Point-biserial associations
SMA_DAT %>%
filter(Measurement_name == "PESop") %>%
summarize(pb_correlation = cor(fois_imp_n, Mean, use="pairwise.complete.obs"),
n = sum(!is.na(Mean)),
p = cor.test(fois_imp_n, Mean)$p.value) %>%
kable(digits = 2)| pb_correlation | n | p |
|---|---|---|
| -0.04 | 65 | 0.76 |
Plots:
SMA_DAT %>%
filter(Measurement_name == "PESop") %>%
ggplot(aes(x = fois_imp_n, y = Mean)) +
geom_point(position = position_jitter(width = .05)) +
geom_smooth(method = "lm", se = F) +
theme_classic()## Warning: Removed 3 rows containing non-finite outside the scale range
## (`stat_smooth()`).## Warning: Removed 3 rows containing missing values or values outside
## the scale range (`geom_point()`).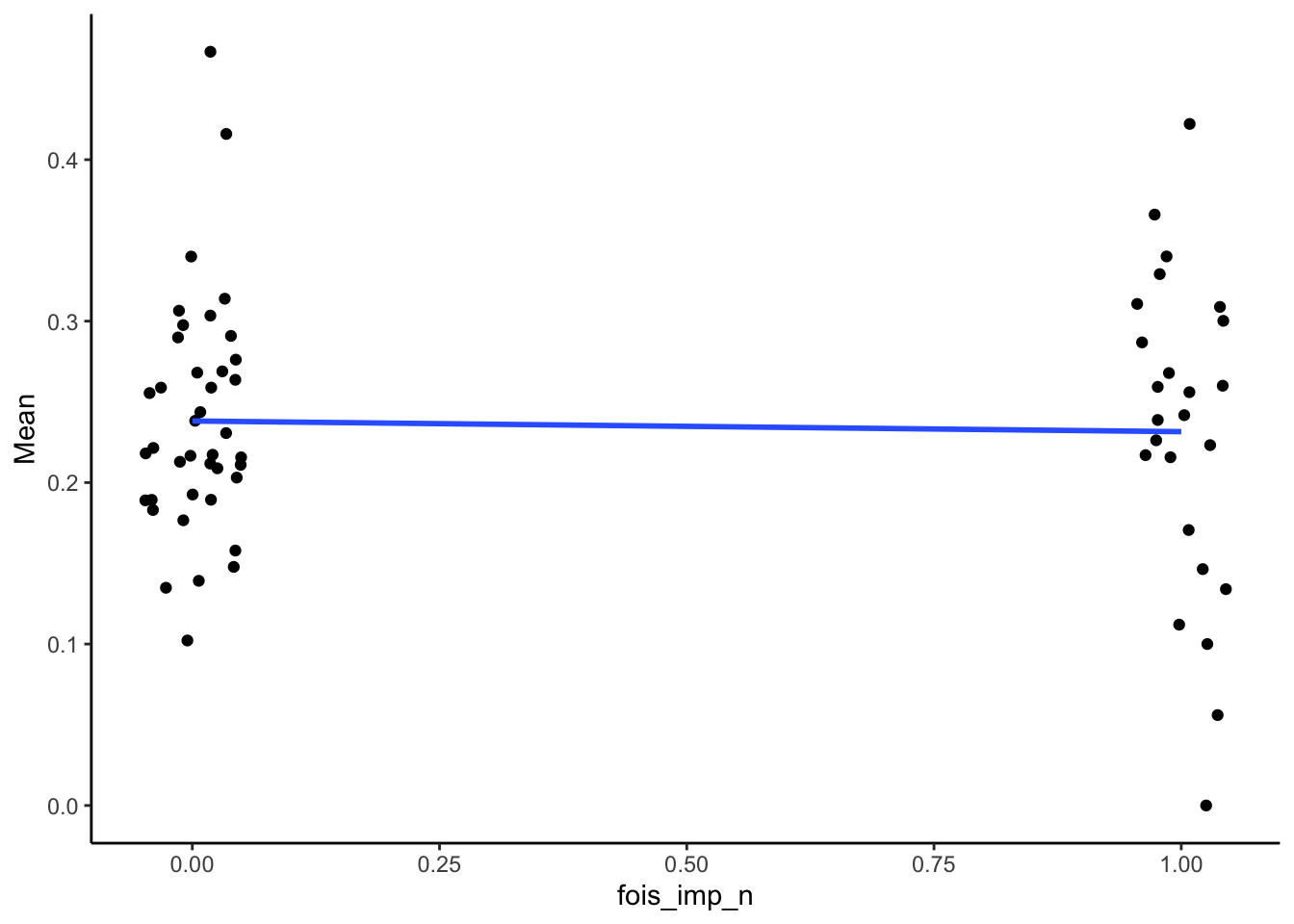
Logistic regressions
foispesop_logr <- glm(fois_imp_n ~ Mean, family = "binomial", data = filter(SMA_DAT, Measurement_name == "PESop"))
foispesop_logr %>%
tidy() %>%
kable(digits = 3)| term | estimate | std.error | statistic | p.value |
|---|---|---|---|---|
| (Intercept) | -0.238 | 0.774 | -0.308 | 0.758 |
| Mean | -0.986 | 3.123 | -0.316 | 0.752 |
NS
Respiratory Support
Point-biserial associations
SMA_DAT %>%
filter(Measurement_name == "PESop") %>%
summarize(pb_correlation = cor(resp_imp_n, Mean, use="pairwise.complete.obs"),
n = sum(!is.na(Mean)),
p = cor.test(resp_imp_n, Mean)$p.value) %>%
kable(digits = 2)| pb_correlation | n | p |
|---|---|---|
| 0.04 | 65 | 0.73 |
Plots:
SMA_DAT %>%
filter(Measurement_name == "PESop") %>%
ggplot(aes(x = resp_imp_n, y = Mean)) +
geom_point(position = position_jitter(width = .05)) +
geom_smooth(method = "lm", se = F) +
theme_classic()## Warning: Removed 3 rows containing non-finite outside the scale range
## (`stat_smooth()`).## Warning: Removed 3 rows containing missing values or values outside
## the scale range (`geom_point()`).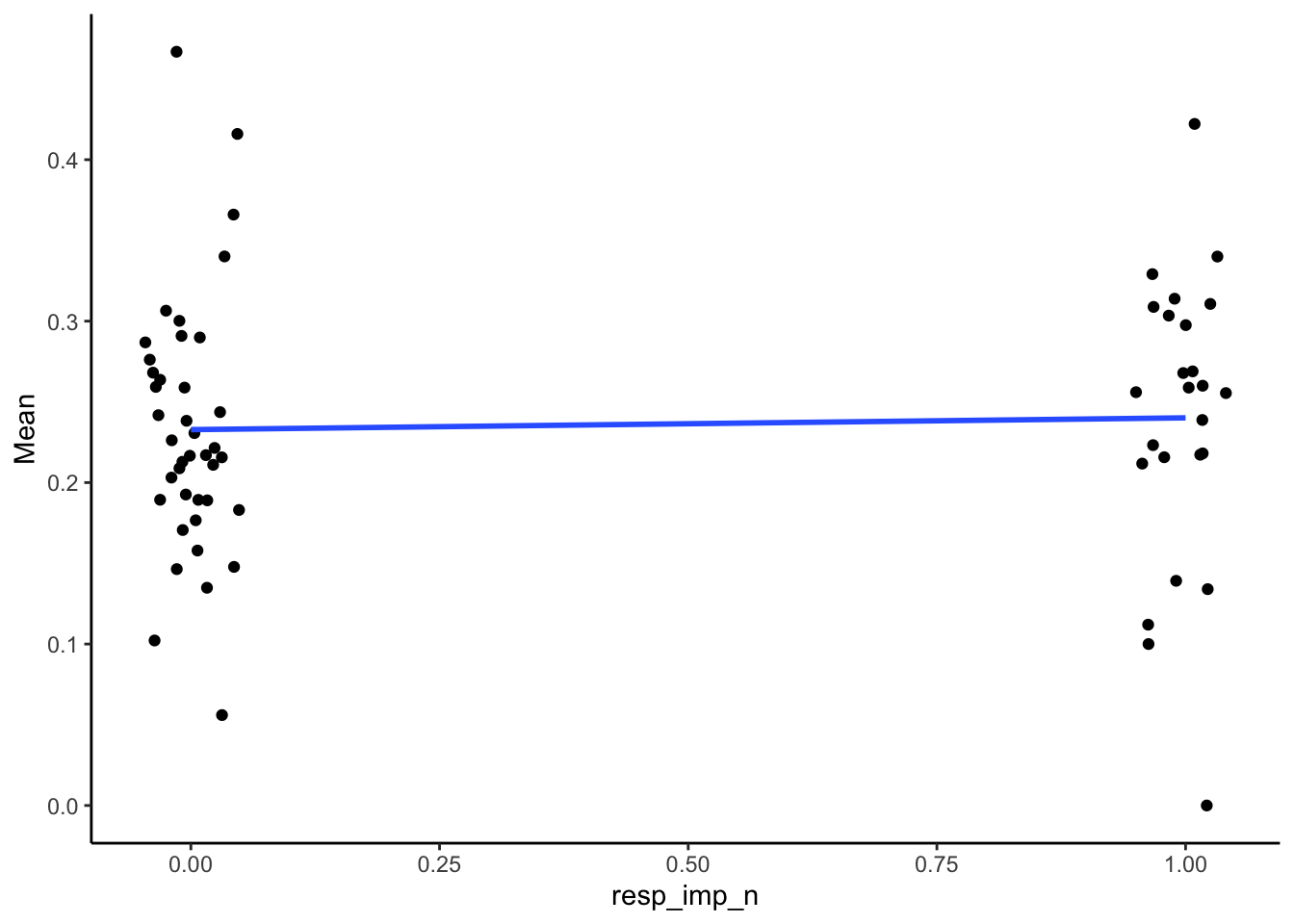
Logistic regressions
resppesop_logr <- glm(resp_imp_n ~ Mean, family = "binomial", data = filter(SMA_DAT, Measurement_name == "PESop"))
resppesop_logr %>%
tidy() %>%
kable(digits = 3)| term | estimate | std.error | statistic | p.value |
|---|---|---|---|---|
| (Intercept) | -0.728 | 0.784 | -0.929 | 0.353 |
| Mean | 1.091 | 3.122 | 0.349 | 0.727 |
NS
Session Information
sessionInfo()## R version 4.5.1 (2025-06-13)
## Platform: x86_64-apple-darwin20
## Running under: macOS Sonoma 14.4.1
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.5-x86_64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.5-x86_64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.1
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: America/Chicago
## tzcode source: internal
##
## attached base packages:
## [1] grid stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] DT_0.33 kableExtra_1.4.0 psych_2.5.6 broom_1.0.8
## [5] effectsize_1.0.1 effsize_0.8.1 nptest_1.1 lme4_1.1-37
## [9] Matrix_1.7-3 irr_0.84.1 lpSolve_5.6.23 irrCAC_1.0
## [13] purrr_1.0.4 pander_0.6.6 DescTools_0.99.60 knitr_1.50
## [17] vcd_1.4-13 readxl_1.4.5 tidyr_1.3.1 ggplot2_3.5.2
## [21] dplyr_1.1.4
##
## loaded via a namespace (and not attached):
## [1] Rdpack_2.6.4 mnormt_2.1.1 gld_2.6.7 rlang_1.1.6
## [5] magrittr_2.0.3 e1071_1.7-16 compiler_4.5.1 mgcv_1.9-3
## [9] systemfonts_1.2.3 vctrs_0.6.5 stringr_1.5.1 pkgconfig_2.0.3
## [13] fastmap_1.2.0 backports_1.5.0 labeling_0.4.3 utf8_1.2.6
## [17] rmarkdown_2.29 tzdb_0.5.0 haven_2.5.5 nloptr_2.2.1
## [21] xfun_0.52 cachem_1.1.0 jsonlite_2.0.0 parallel_4.5.1
## [25] R6_2.6.1 bslib_0.9.0 stringi_1.8.7 RColorBrewer_1.1-3
## [29] boot_1.3-31 lmtest_0.9-40 jquerylib_0.1.4 cellranger_1.1.0
## [33] estimability_1.5.1 Rcpp_1.0.14 zoo_1.8-14 parameters_0.27.0
## [37] pacman_0.5.1 readr_2.1.5 splines_4.5.1 tidyselect_1.2.1
## [41] rstudioapi_0.17.1 yaml_2.3.10 lattice_0.22-7 tibble_3.3.0
## [45] withr_3.0.2 bayestestR_0.16.1 coda_0.19-4.1 evaluate_1.0.4
## [49] proxy_0.4-27 xml2_1.3.8 pillar_1.10.2 reformulas_0.4.1
## [53] insight_1.3.1 generics_0.1.4 hms_1.1.3 scales_1.4.0
## [57] rootSolve_1.8.2.4 minqa_1.2.8 xtable_1.8-4 class_7.3-23
## [61] glue_1.8.0 emmeans_1.11.1 lmom_3.2 tools_4.5.1
## [65] data.table_1.17.6 forcats_1.0.0 Exact_3.3 fs_1.6.6
## [69] mvtnorm_1.3-3 rbibutils_2.3 crosstalk_1.2.1 datawizard_1.1.0
## [73] colorspace_2.1-1 nlme_3.1-168 cli_3.6.5 textshaping_1.0.1
## [77] expm_1.0-0 viridisLite_0.4.2 svglite_2.2.1 gtable_0.3.6
## [81] sass_0.4.10 digest_0.6.37 htmlwidgets_1.6.4 farver_2.1.2
## [85] htmltools_0.5.8.1 lifecycle_1.0.4 httr_1.4.7 MASS_7.3-65